
")

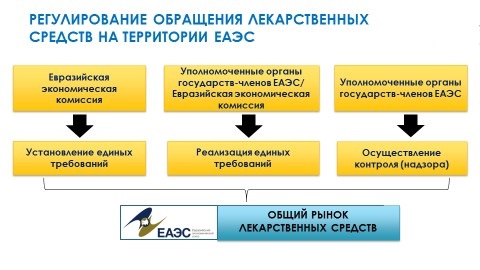
Основу нормативно-правового регулирования обращения лекарственных средств на едином рынке составляет:
- базовый документ «первого уровня» Соглашение о единых принципах и правилах обращения лекарственных средств в рамках ЕАЭС от 23 декабря 2014 года;
- 26 документов второго уровня, которые разработаны и уже утверждены;
- 68 нормативно-правовых актов третьего уровня, работа над которыми ведется;
- а также национальное законодательство государств-членов ЕАЭС, которое должно быть приведено в соответствие с требованиями законодательства Союза.
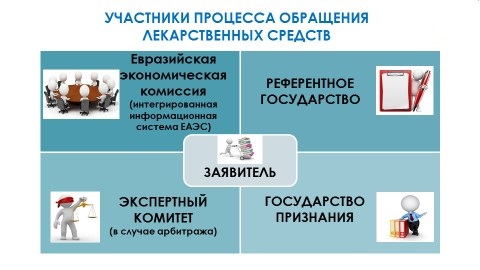
Основным участником процесса регистрации лекарственных средств на едином рынке ЕАЭС является заявитель, который выбирает референтное государство и государство признания. Эти три субъекта взаимодействуют между собой в рамках информационной интегрированной системы Евразийского экономического союза. Все возникающие в ходе регистрации вопросы и несогласия рассматриваются Экспертным комитетом.
В чем преимущества общего рынка лекарственных средств?
Самое главное, это обеспечение единых подходов и требований к качеству, безопасности и эффективности лекарственных средств. При этом за основу взят самый лучший мировой опыт:
- проведение доклинических и клинических исследований в соответствии с надлежащими практиками;
- производство лекарственных препаратов в условиях GMP;
- признание результатов клинических, доклинических испытаний, проведенных в соответствии с правилами GLP, GCP;
- признание инспекций надлежащих фармацевтических практик в рамках Союза;
- снижение роста непроизводительных затрат и повышение экономической доступности лекарственных препаратов;
- создание конкурентоспособных отечественных лекарственных препаратов (под отечественными следует понимать лекарственные средства, произведенные на территории всех стран-членов ЕАЭС);
- увеличение рынка сбыта лекарственных препаратов с выходом на зарубежные рынки.
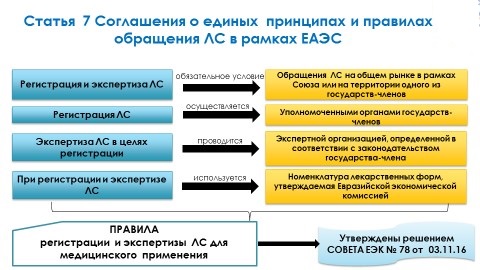
Для обращения ЛС на общем рынке в рамках Союза или на территории одного из государств-членов обязательным условием является его регистрация и экспертиза. Регистрация осуществляется уполномоченными органами государств-членов Союза. Экспертные органы определяются законодательством стран-членов и при проведении экспертизы они руководствуются нормативно-правовыми актами, разработанными Рабочими группами ЕАЭС и утвержденными Советом ЕЭК.
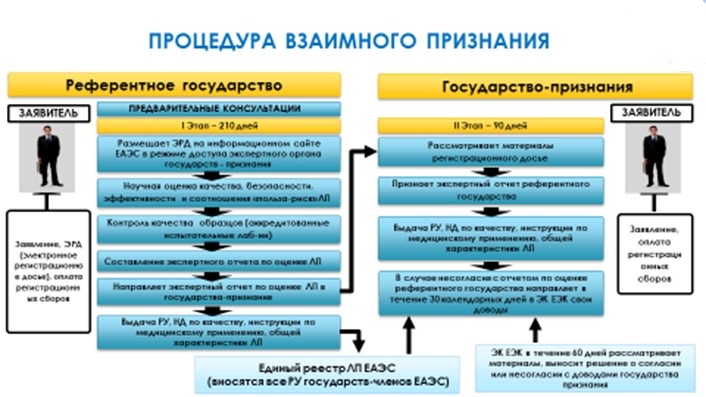
Система допуска лекарственных средств состоит из трех процедур.
Процедура взаимного признания – это два последовательных этапа:
1) этап регистрации в референтном государстве, проведение экспертизы регистрационного досье, проведение лабораторных испытаний ЛП, при необходимости проведение внеплановых инспекций GxP (210 календарных дней);
2) этап признания государствами признания экспертного отчета, составленного референтным государством (90 календарных дней).
Критериями выбора референтного государства являются наличие опыта научной деятельности, готовность защищать досье заявителя, прозрачность и доступность для заявителя, способность соблюдать сроки экспертизы, предшествующий опыт проводимых экспертиз.
При децентрализованной процедуре процесс осуществляется одновременно референтным государством и несколькимигосударствами-членами Союза. Заявитель самостоятельно осуществляет выбор референтного государства при подаче заявления на регистрацию лекарственного средства. После принятия положительного решения наступает период выдачи национальных регистрационных удостоверений, перевод ОХЛП, инструкций, макетов упаковок. При достижении взаимного согласия эта процедура может быть закончена в срок до 210 дней во всех государствах. Основное требование – отсутствие выданных РУ в странах на момент подачи заявки.
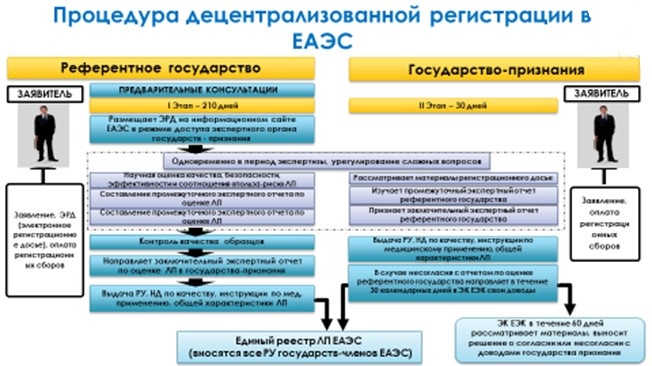
И национальная процедура, когда процесс осуществляется национальными компетентными органами в соответствии с национальным законодательством государства-члена ЕАЭС. Данная процедура будет действовать до 31 декабря 2015 года.
В настоящее время идет работа по разработке прейскуранта цен на регистрацию лекарственных препаратов на территории ЕАЭС.
Регистрационные досье лекарств, зарегистрированных в государствах-членах Союза до вступления в силу Соглашения, должны быть приведены в соответствие с требованиями Союза до 31 декабря 2025 года. Для этого заявитель подает в государство, где зарегистрирован лекарственный препарат, модули 1-3 регистрационного досье, если лекарственный препарат предназначен для обращения на территории государства-члена, в котором он зарегистрирован. Данные доклинических и клинических исследований в этом случае представляются в модулях 4-5 РД в виде соответствующих отчетов. В случае дальнейшей регистрации по процедуре взаимного признания в государствах-членах Союза, в которых данный ЛП не был зарегистрирован до вступления Соглашения в силу, предоставляются модули 1-5 РД.
Государства-члены ЕАЭС в процессе проведения процедур экспертизы осуществляют взаимодействие через интегрированную информационную систему по вопросам переписки с заявителями, направления промежуточных отчетов в государства-признания, окончательного экспертного отчета.Экспертиза ЛП в случае инициирования фармацевтической инспекции на соответствие GxP Союза не приостанавливается и должна быть проведена в срок регистрации ЛП, но не превышающий 180 календарных дней с даты принятия решения об инициировании инспекции.
В процессе экспертизы для получения дополнительной информации от заявителя применяется принцип остановки часов (не более 180 дней), не входящий в общий срок экспертизы.
Регистрационное удостоверение выдается под единым номером сроком на 5 лет с последующим подтверждением регистрации (перерегистрации) на бессрочный период.
По окончании процедуры регистрации утверждаются и выдаются заявителю РУ, общая характеристика ЛП, инструкция по медицинскому применению (листок-вкладыш), макеты упаковок на языке по национальным требованиям государства Союза.
Государство признания при неодобрении экспертного отчета референтного государства обращается в Экспертный комитет для рассмотрения разногласий в соответствии с порядком, установленным ЕЭК.
Экспертный комитет по ЛС создается при Евразийской Экономической Комиссии и формируется Коллегией Комиссии на 3 года по 3 кандидатуры от каждого государства-члена Союза. Он рассматривает вопросы в сфере обращения ЛС путем подготовки рекомендаций по возникающим разногласиям:
- по регистрации и экспертизе;
- признанию результатов ДКИ и КИ, результатов инспектирования;
- расхождению позиций по вопросу соотношения польза/риск ЛС, находящихся в обращении;
- необходимости проведения совместных инспекций GxP;
- выбору референтного препарата для проведения ДКИ и КИ и др.
Решения Экспертного комитета носят рекомендательный характер. Срок рассмотрения обращений – 60 календарных дней.
Итак, основным условием выхода лекарственного препарата на общий рынок ЕАЭС является наличие сертификата GMP Союза (или не ниже). При его отсутствии предоставляется действующий сертификат GMP страны-производителя, копия последнего инспекционного отчета, сведения о результатах всех инспекций за предшествующие три года, сведения о рекламациях, согласие на проведение фармацевтической инспекции, копия мастер-файла. ДКИ и КИ, проведенные в-третьих странах, рассматриваются в процессе экспертизы при условии, что они спланированы, проведены в соответствии с требованиями GLP, GCP, эквивалентных требованиям Союза (или не ниже). Лабораторные испытания образцов ЛП осуществляются референтным государством в аккредитованных лабораториях государства или в определенных случаях в лабораториях производителей. Образцы ЛП и стандартные образцы предоставляются только в референтное государство. Регистрационное досье предоставляется на электронном носителе (дополнительно модуль 1 на бумажном носителе в соответствии с национальными требованиями) с доступом всех государств признания через интегрированную информационную систему к данным РД.
До полноценной работы общего рынка лекарств на территории всех государств-членов ЕАЭС предусмотрен переходный период. Так, ЛС, зарегистрированные в государствах-членах Союза до вступления в силу Соглашения, должны быть приведены в соответствие с требованиями и правилами Союза до 31 декабря 2025 г. Государства-члены допускают подтверждение регистрации ЛС, имеющих срочные РУ, выданные до вступления в силу Соглашения, по истечении их срока действия в соответствии с национальным законодательством государств-членов Союза. Регистрация лекарственного препарата, поданного на регистрацию до 31 декабря 2020 г., может быть осуществлена в соответствии с законодательством государства-члена без учета требований Правил Союза (пункт 184 Правил ЕАЭС).
Подготовила Гульжамал Раисова.







